
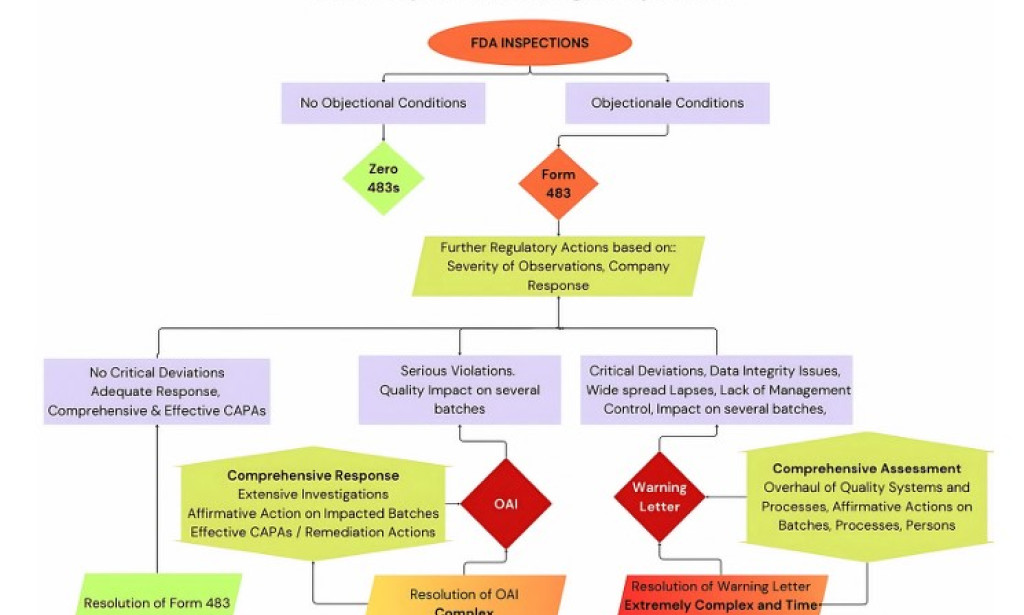
Of particular concern are FDA 483s issued mainly by the U.S. Food and Drug Administration (FDA) for violations of current Good Manufacturing Practices (cGMP) Cosmetics facility design consultants in India, data integrity lapses, inadequate quality controls and associated issues. Such letters have serious implications — affecting market access, reputation, and compliance costs.
COMMON THEMES / SYSTEMIC PROBLEMS
Majority of issues of getting the warning letters are lies with the governing principles of the manufacturing companies and some of them are highlighted below after studying few of warning letters issued to various Indian pharma manufacturing companies in year 2025
Poor / Incomplete Quality Control Units (QC Units)
Several warning letters highlight QC units failing to provide adequate oversight, failing to test incoming raw materials & maintaining records, lacking suitable control over testing and procedures.
Data Integrity Issues
Manipulated or falsified data, inadequate documentation, limited or blocked access to records, gaps in laboratory documentation.
Cleaning / Equipment / Cross‐contamination Controls
Deficient cleaning procedures, lack of clarity on cleaning validation, prevention of contamination or cross‑contamination
Stability Program Deficiencies
Delays in testing, backlog of stability data (i.e. products already released without full stability data), improper or inconsistent stability program.
Lack of Transparency / Obstruction during Inspections
In at least one case, access was limited or initially denied, and documents were not fully made available
Contract Manufacturing Oversight
Where parts of production are outsourced, oversight has been weak or absent, with companies being held responsible for issues arising in contract facilities.
IMPLICATIONS OF THESE WARNING LETTERS
These warning letters carry consequences that go beyond just internal corrective actions. Here are some of the key effects:
Import Alerts / Market Access Restriction
Companies under warning letters often find themselves on Import Alerts (such as Import Alert 66-40), which means FDA may refuse their shipments into the U.S and imported products are subject to automatic detention ( DWPE), until compliance issues are addressed. This threatens revenues and market presence
Delays in New Product Approvals
Facilities flagged for violations may see delays in new product approvals or filings, especially if those facilities are responsible for the manufacturing of those products.
Reputational Damage
Being publicly named in FDA warning letters erodes trust among buyers (both domestic and international), investors and regulatory bodies. This could affect future business opportunity and partnerships.
Cost of Remediation
Corrective and preventive actions (CAPA) require investment: Upgrading facilities, better documentation systems, more stringent QC, training of staff, possibly revalidating processes. These costs may be high, especially for smaller companies
Regulatory Scrutiny & Oversight Pressure
These incidents increase scrutiny from regulators (both Indian and abroad). Indian regulators (like CDSCO) may be pressured to tighten oversight and enforcement. Companies might have to prepare for more frequent inspections or stricter compliance rules
Supply Chain Disruption
If a facility is non‑compliant and imports are refused, it can affect availability of medicines, or force companies to switch to alternate manufacturing units, possibly at higher cost

You must be logged in to post a comment.